Coming out of the (Bayesian) closet: multivariate version
This week I’m facing my—and many other lecturers’—least favorite part of teaching: grading exams. In a supreme act of procrastination I will continue the previous post, and the antepenultimate one, showing the code for a bivariate analysis of a randomized complete block design.
Just to recap, the results from the REML multivariate analysis (that used ASReml-R) was the following:
library(asreml)
m4 <- asreml(cbind(bden, veloc) ~ trait,
random = ~ us(trait):Block + us(trait):Family, data = a,
rcov = ~ units:us(trait))
summary(m4)$varcomp
# gamma component std.error
#trait:Block!trait.bden:bden 1.628812e+02 1.628812e+02 7.854123e+01
#trait:Block!trait.veloc:bden 1.960789e-01 1.960789e-01 2.273473e-01
#trait:Block!trait.veloc:veloc 2.185595e-03 2.185595e-03 1.205128e-03
#trait:Family!trait.bden:bden 8.248391e+01 8.248391e+01 2.932427e+01
#trait:Family!trait.veloc:bden 1.594152e-01 1.594152e-01 1.138992e-01
#trait:Family!trait.veloc:veloc 2.264225e-03 2.264225e-03 8.188618e-04
#R!variance 1.000000e+00 1.000000e+00 NA
#R!trait.bden:bden 5.460010e+02 5.460010e+02 3.712833e+01
#R!trait.veloc:bden 6.028132e-01 6.028132e-01 1.387624e-01
#R!trait.veloc:veloc 1.710482e-02 1.710482e-02 9.820673e-04
# z.ratio constraint
#trait:Block!trait.bden:bden 2.0738303 Positive
#trait:Block!trait.veloc:bden 0.8624639 Positive
#trait:Block!trait.veloc:veloc 1.8135789 Positive
#trait:Family!trait.bden:bden 2.8128203 Positive
#trait:Family!trait.veloc:bden 1.3996166 Positive
#trait:Family!trait.veloc:veloc 2.7650886 Positive
#R!variance NA Fixed
#R!trait.bden:bden 14.7057812 Positive
#R!trait.veloc:bden 4.3442117 Positive
#R!trait.veloc:veloc 17.4171524 Positive
The corresponding MCMCglmm code is not that different from ASReml-R, after which it is modeled anyway. Following the recommendations of the MCMCglmm Course Notes (included with the package), the priors have been expanded to diagonal matrices with degree of belief equal to the number of traits. The general intercept is dropped (-1) so the trait keyword represents trait means. We are fitting unstructured (us(trait)) covariance matrices for both Block and Family, as well as an unstructured covariance matrix for the residuals. Finally, both traits follow a gaussian distribution:
library(MCMCglmm)
bp <- list(R = list(V = diag(c(0.007, 260)), n = 2),
G = list(G1 = list(V = diag(c(0.007, 260)), n = 2),
G2 = list(V = diag(c(0.007, 260)), n = 2)))
bmod <- MCMCglmm(cbind(veloc, bden) ~ trait - 1,
random = ~ us(trait):Block + us(trait):Family,
rcov = ~ us(trait):units,
family = c('gaussian', 'gaussian'),
data = a,
prior = bp,
verbose = FALSE,
pr = TRUE,
burnin = 10000,
nitt = 20000,
thin = 10)
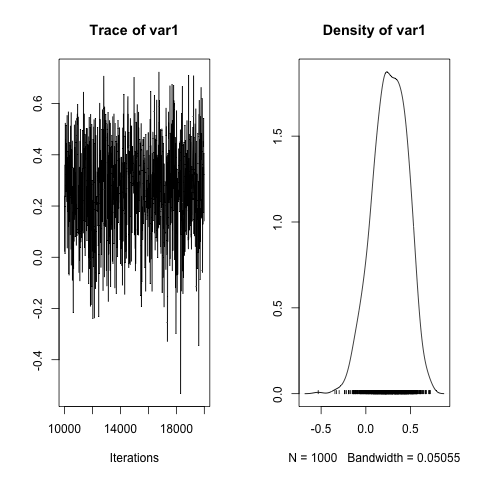
Further manipulation of the posterior distributions requires having an idea of the names used to store the results. Following that, we can build an estimate of the genetic correlation between the traits (Family covariance between traits divided by the square root of the product of the Family variances). Incidentally, it wouldn’t be a bad idea to run a much longer chain for this model, so the plot of the posterior for the correlation looks better, but I’m short of time:
dimnames(bmod$VCV)
rg <- bmod$VCV[,'veloc:bden.Family']/sqrt(bmod$VCV[,'veloc:veloc.Family'] *
bmod$VCV[,'bden:bden.Family'])
plot(rg)
posterior.mode(rg)
# var1
#0.2221953
HPDinterval(rg, prob = 0.95)
# lower upper
#var1 -0.132996 0.5764006
#attr(,"Probability")
#[1] 0.95
And that’s it! Time to congratulate Jarrod Hadfield for developing this package.